July
9
Antibiotic Stock, LB and LB + chloramphenicol plate preparation- 34 mg/mL chloramphenicol stock solution is prepared by adding 0.378 g of 900 mg/g chloramphenicol stock in 10 mL of 95% ethanol. The stock solution is then distributed into 10 microfuge tubes as 1 mL aliquots.
- 1 L of LB is prepared for experimental use. In order to prepare 1 L of LB, 1 L of water is measured in a graduated cylinder and then is added into a flask. 25 g of solid LB broth is added to the flask along with a stir bar. The flask is placed on a magnetic stirrer to get mixed thoroughly. The mixed solution then is distributed into 5 different glass bottles as 200 mL aliquots. These bottles are then autoclaved by using liquid 45 cycle.
- 300 mL of LB is also prepared along with other solutions. 300 mL of distilled water is measured with a graduated cylinder and then is transferred into a flask. 7.5 g of solid LB broth is added to the flask along with a stir bar. The flask is placed on a magnetic stirrer to get mixed thoroughly. The solution is then autoclaved by using liquid 45 cycle.
- After the autoclaving process, the flask containing 300 mL of LB is cooled down in a 50 C incubator. Once the solution is cooled down, 300 μL of cold 34 mg/mL stock solution is added to have a final [chloramphenicol] of 34 μg/mL. The solution is mixed for a minute in a magnetic stirrer and poured into empty sterile agar plates. No flame was used in this process as the gas line of the lab was problematic. After 20 minutes, the plates were stacked into a sterile plate bag and were kept in a cold room (4C) for further use.
24
Sterilization of Tubes for experimental use and additional preparation of antibiotic plates- Pipette tips, 12 mL test tubes, microfuge tubes, PCR tubes, glycerol stock tubes and wooden toothpicks were autoclaved using a Gravity Filtration Cycle.
- 2 different 300 mL of LB is prepared in order to pour out kanamycin (50 μg/mL) and chloramphenicol (34 μg/mL) plates. 300 mL of distilled water is measured with a graduated cylinder and then is transferred into a flask. 7.5 g of solid LB broth is added to the flask along with a stir bar. The flask is placed on a magnetic stirrer to get mixed thoroughly. Each flask is covered by a thin foil and the solutions are then autoclaved by using the liquid 45 cycle.
- 50 mL of 100% glycerol is measured by a graduated cylinder and then is added into a 150 mL glass bottle containing 50 mL of milli-q Water to have a 50% glycerol stock solution. The solution is then autoclaved with LB solutions.
- After the autoclaving process, the flask containing 300 mL of LB is cooled down in a 50 C incubator. Once the solution is cooled down, 300 μL of cold 34 mg/mL stock solution is added to have a final [chloramphenicol] of 34 μg/mL. The solution is mixed for a minute in a magnetic stirrer and is aseptically poured into empty sterile agar plates. After 20 minutes, the plates were stacked into a sterile plate bag and were kept in a cold room (4C) for further use. The same principle is used to pour out kanamycin plates but 300 μL of kanamycin (50mg/mL) stock solution is used.
25
Calibration Experiment for the 2022 iGEM Interlab Study- The calibration experiment for the Interlab study was conducted based on the protocol provided by iGEM. As the wavelength values of the Plate Reader cannot be modified, the measurement is left to be done later on.
26
Primer dilutions- 100 mM primer dilutions are done by adding 10X volume (μL) of the nmole value of each primer. Primer stocks are vortexed vigorously both before and after the addition of corresponding volume of sterile mq water. 10 mM primer dilutions are done for the primers #35-42 by adding 10 μL of each 100 mM primer dilution into a sterile microfuge tube along with 90 μL sterile mq water. All stock solutions are kept in -20 freezer for further use
27
Colony PCR to amplify HPA loci containing hpaA and hpaX along with their corresponding promoters- PCR calculations are done in advance
- Two single, well-isolated E.coli BL21 colonies are picked by using a sterile tip + a P20 pipette and are added into two different microfuge tubes. These colonies are resuspended by adding 20 μL of sterile mq water
- Corresponding amount of mq water, 10X PCR Buffer, MgSO4 , 5 U/μL of Platinum Taq polymerase and 2.5 U/μL of Pfu polymerase are added respectively into a microfuge tube to create a master mix
- 3.5 μL of each primer (forward and reverse) are added into a separate microfuge tube for each PCR reaction. 61 μL of master mix then is added into each tube and mixed thoroughly by pipetting up and down
- 2 μL of each microfuge tube corresponding to resuspended tubes are transferred into 3 different tubes and are labeled as Reaction 1 Colony 1, Reaction 2 Colony 1, NC Colony 1 and Reaction 2 Colony 1, Reaction 2 Colony 2, NC Colony 2. 18.5 μL of the corresponding master mix is added into each PCR tube and mixed carefully by pipetting up and down. Corresponding settings are set-up in the Thermocycler (name the brand) by using the designated configurations : 4 minutes of initial denaturation at 96 C, a total of 30 cycles of 15 seconds of denaturation at 96 C + 30 seconds of annealing at 58 degrees (the annealing temperature is calculated by extracting 3 C from the annealing temperature of the primer with the lowest Tm value) and 60 seconds of extension step at 72 C. A final extension step is added for 5 minutes at 72C to make sure that all the products are indeed full-sized products. Reaction tubes are kept in -20 C freezer for further use.
- Also both 1 and 2% agarose gels were prepared and PCR products were run on these gels. The fragment #2 was successfully amplified as we observed a band around 500 bp yet no expected amplification occurred for the fragment #1. Please check out Joseph’s gel results.
28
Test transformations for troubleshooting- 1.25 uL of resuspended pJUMP27-1A plasmid (BBa_J428351) and one of the Interlab STudy # 2 plasmids were added into two different tubes each containing 50 uL of competent Dh5alpha cells. The protocol of transformation by chemical competence is exactly followed. (The cells were kept on ice for an hour instead of 30 minutes.) The cells were heat shocked at 47 C due to instrument limitations and this step probably caused a significant decrease in cell viability. As the lab did not contain a plate spreader, the cells were plated onto LB + CAM 34 plates using a metal spatula. These were grown overnight at 37 C. No colonies were observed the very next day.
- Heat shock was the most likely reason why the transformations failed. Yet, the amount of DNA that is added into each transformation reaction is not likely to be sufficient. From my experience, the lowest [DNA] that I used was ~ 20 ng/uL and the transformations worked perfectly fine even in that case. Yet, the maximum DNA that can be added is 3 ng which would not be sufficient for a successful transformation reaction.
29
Test transformations for troubleshooting (again)- This time 1, 3 and 5 uL of the resuspended plasmid (check out the exact plasmid) were added separately into 3 different microfuge tubes containing 50 uL of DH5alpha comp cells. The transformation by chemical competence protocol is exactly followed without making any changes. At the end, each pelleted and resuspended cells were plated onto LB+ CAM 34 plates and these were grown overnight at 37 C. No growth was observed the next day. Once again, the most likely reason for failed transformations is the insufficient [DNA]. There might be another possibility that the [CAM] is wrong or it is too much for successful cell growth.
- There should not be any problem with the transformation protocol. I ran around 40 different transformation reactions with this exact protocol and in every case there was at least 25 colonies. The most likely problem is the amount of DNA added is simply insufficient for a successful transformation
30
Test transformations for troubleshooting (again)- In this case 4 uL of resuspended plasmid (BBa_J23100) is added into two microfuge tubes containing 100 uL of DH5alpha comp cells. Also, 8 uL of resuspended DNA (BBa_I20270) is added into a different tube with 100 uL of comp cells. The transformation by chemical competence protocol is exactly followed without making any changes. The resuspended pellet of cells with 4 uL of resuspended pellet were plated onto one LB and one LB + CAM 34 plate. Cells containing 8 uL of DNA were pelleted and resuspended. 80 uL of the resuspension were plated onto two different plates (LB and LB+ CAM)
31
Colony PCR to amplify HPA loci containing hpaA and hpaX along with their corresponding promoters and Pbc promoter- 5-6 colonies were observed in the 13A LB+CAM 34 plate (the first successful transformation) whereas no growth was observed in 13B LB+CAM 34 plate
- There was an immense growth in each LB plate. It was a bad idea to plate everything in LB plates as well. This was done in order to see whether there was a problem with [CAM] in our plates. Eventually, all the cells, regardless of the success of transformation, grew on LB plates. What I should have done was to plate out transformants into different [CAM] to see whether a change in concentration might result in a difference.
-
The PCR reaction is done based on this calculations and instructions:
Component [Stock] [Final] 1 Reaction Master Mix (x10) Sterile mqWater - - 14.93 149.3 PCR Buffer 10X 1X 2.5 μL 25 μL MgSO4 25 mM 2.5 mM 2.5 μL 25 μL dNTPs 25 mM 0.4 mM 0.4 μL 4.0 μL Taq Pol 5 U/μL 0.725 U/rxn 0.14 μL 1.4 μL Pfu 2.5 U/μL 0.08 U/rxn 0.03 μL 0.3 μL Mix total - - 20.5 205 Template (cell suspension) - - 2.0 μL - Final volume - - 25.0 μL - Fwd primer 10 uM 0.5 uM 1.25 4.375 Rev primer 10 uM 0.5 uM 1.25 4.375 - Add 4.375 uL of fwd and rev primers into 3 different microfuge tubes and add 69 μL master mix into those -> distribute into PCR tubes
- 6 amplification reactions + 3 negative controls
-
Initial denaturation 4 minutes at 96
Denaturation 15 seconds at 96
Annealing 30 seconds at 59 for normal primers and 52.5 for Pbc primers
Elongation 90 seconds at 72 for HPA amplification, 45 seconds for Pbc
Final extension 5 minutes at 72
Hold 10 degrees - X 30 cycles of denaturation + annealing + elongation
- One 1% and one 1.5% of agarose gels were prepared and the products were run in these gels. The bands were not really thick, the most likely reason is that there wasn’t sufficient EtBr in the TAE buffer. Yet, it seems like we successfully amplified fragment 2 of HPA and Pbc promoter based on fragment sizes.
- The fragment #1 of HPA still does not work. There is likely to be a problem with the primers. The ™ difference between those two is around 3-5 which should be OK. None of those primers would give primer dimer amplification as the major product. Yet, it seems like these two primers can also anneal with themselves to a certain extent. We might want to ask Dr.Stuart about what can be done in order to increase the PCR efficiency. In my opinion, we can use DMSO to increase the annealing specificity of the primers to the desired DNA region.
- NOTE : Based on August 1’s results, it seems like the major problem is the cross-dimer of rev and primers so reducing the primer concentration might be the first thing to do.
- I also realized that I have adding slightly more than 0.5 uM of primers
August
1
Test transformations to see whether [chloramphenicol] is the problem- Poulomi can provide the details
2
Restreaking colonies onto a LB + Cam 34 plate + Overnight culture preparation for E.coli BL21- Two colonies are observed in Cam 15 plate. One of the colonies is picked aseptically with a toothpick and restreaked onto a new CAM 34 plate. A significant number of colonies were observed the other day. Based on these observations, we concluded that the [antibiotic] is not the reason for low transformation efficiency.
- An overnight culture of BL21 is prepared in 5 mL of LB. The colony is picked and dumped into the tube aseptically. A negative control of LB + no colony is also prepared to see if the LB is still sterile. Only growth is observed for the LB+colony tube indicating that the LB is not contaminated. From this tube, a glycerol stock of BL21 is prepared by adding 500 uL of the overnight culture into 500 uL of sterile 50% glycerol
3
PCR optimization for HPA fragment #1 and getting more PCR products of HPA #2 for purification-
Component [Stock] [Final] 1 Reaction Master Mix (x15) Sterile mqWater - - 13.93 μL 208.95 μL PCR Buffer 10X 1X 2.5 μL 37.5 μL MgSO4 25 mM 2.5 mM 2.5 μL 37.5 μL dNTPs 25 mM 0.4 mM 0.4 μL 6.0 μL Taq Pol 5 U/mL 0.725 U/reaction 0.14 μL 2.1 μL Pfu 2.5 U/mL 0.08 U/reaction 0.03 μL 0.45 μL Mix total - - 19.5 μL 292.5 μL Template (cell suspension) - - 3.0 μL - Final volume - - 25.0 μL - Component [Stock] [Final] 1 Reaction Master Mix (x2.143) NC purposes Extra mq water to add to 25 uL (x1)
Extra mq water to add to 25 uL (x2.143)
Fwd primer 10 μM
0.5 μM
1.25 2.68 Add 4.02 - - Rev primer 10 μM
0.5 μM
1.25 2.68 Add 4.02 - - Fwd primer 10 μM
0.3 μM
0.75 1.61 - 1 2.14 Rev primer 10 μM
0.3 μM
0.75 1.61 - - Fwd primer 10 μM
0.15 μM
0.375 0.8 - 1.75 3.75 Rev primer 10 μM
0.15 μM
0.375 0.8 - 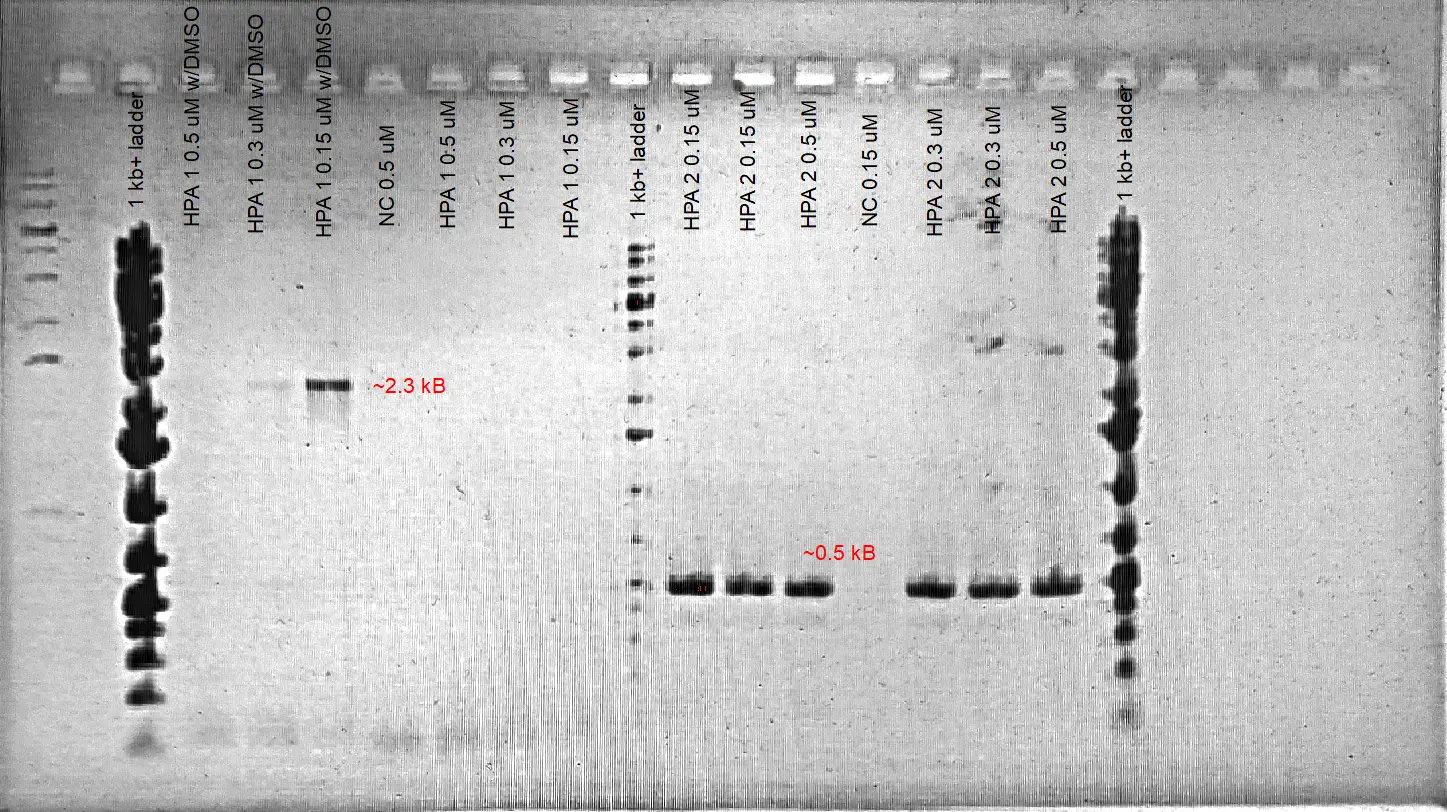
1% Agarose Gel Electrophoresis Results of HPA Fragments #1 and #2.
4
Gradient PCR for HPA Fragment 1 + Final Test Transformations-
Gradient PCR for the HPA fragment #1 is tested in order to increase product specificity. We also added different [final] DMSO (%3 and 5%) into separate reactions to determine whether it might also increase the product efficiency.
- 5% DMSO increases the product yield as it is likely to increase primer binding specificity to the template. Annealing temperature values of 61 and 58.7 C resulted in the amplification of the desired product when 3-5 %DMSO is also added. No amplification of the expected fragment is observed when the annealing temperature is dropped to 55 C. The desired fragment is obtained around 2.3 kB which is the expected product size.
6
- Prepared 1L of LB for experimental use
- Colony PCR for the amplification of pBC promoter from E.coli BL21
-
Run a gradient PCR with the respective ™ values : 52.5 C and 54 C
4 amplification reactions + 1 negative controlComponent [Stock] [Final] 1 Reaction Master Mix (x6) Sterile mqWater - - 14.92 μL 89.537 μL PCR Buffer 10X 1X 2.5 μL 15 μL MgSO4 25 mM 2.5 mM 2.5 μL 15 μL dNTPs 25 mM 0.4 mM 0.4 μL 2.4μL Taq Pol 5 U/mL 0.029 U/μL 0.145 μL 0.87 μL Pfu 2.5 U/mL 0.0032 U/μL 0.032 μL 0.193 μL Fwd Primer 10 μM 0.3 μM 0.75 μL 4.5 μL Rev Primer 10 μM 0.3 μM 0.75 μL 4.5 μL Mix total - - 22 μL 132 μL Template (cell suspension) - - 3.0 μL - Final volume - - 25.0 μL -
Initial denaturation 4 minutes at 96
Denaturation 15 seconds at 96
Annealing 30 seconds at 52.5 /54 C
Elongation 45 seconds at 72
Final extension 5 minutes at 72
Hold 10 C - Denaturation + Annealing + Elongation steps are repeated for 30 cycles
- ON culture preparation of the two transformed plasmids to do minipreps. This is being done in order to observe fina [plasmid] in cells. Cultures are aseptically inoculated into separate tubes containing 5 mL of LB with a toothpick.
7
-
Gradient PCR to amplify the GFP + B0015 terminator sequence from the iGEM plasmid BBa_J364001 (™ values of 56 and 53 C are used for the PCR) 1 uL of DNA is added into each tube whereas 1 uL of sterile mqwater is added to the control tube.
Component [Stock] [Final] 1 Reaction Master Mix (x4) Sterile mqWater - - 16.92 μL 67.69 μL PCR Buffer 10X 1X 2.5 μL 10 μL MgSO4 25 mM 2.5 mM 2.5 μL 10 μL dNTPs 25 mM 0.4 mM 0.4 μL 1.6 μL Taq Pol 5 U/mL 0.029 U/μL 0.145 μL 0.58 μL Pfu 2.5 U/mL 0.0032 U/μL 0.032 μL 0.129 μL Fwd Primer 10 μM 0.3 μM 0.75 μL 3.0 μL Rev Primer 10 μM 0.3 μM 0.75 μL 3.0 μL Mix total - - 24 μL 96 μL Template (GFP containing plasmid)
- - 1.0 μL - Final volume - - 25.0 μL - -
4 amplification reactions + 1 negative control
Initial denaturation 4 minutes at 96 (could have used 2 minutes)
Denaturation 15 seconds at 96
Annealing 30 seconds at 53 /56 C
Elongation 75 seconds at 72
Final extension 5 minutes at 72
Hold 10 C - Denaturation + Annealing + Elongation steps are repeated for 30 cycles
-
1% gel electrophoresis is run in order to determine product sizes
Note : Gel image is aborted as it didn’t provide corresponding details
8
1% Gel electrophoresis for the PCR products of GFP-
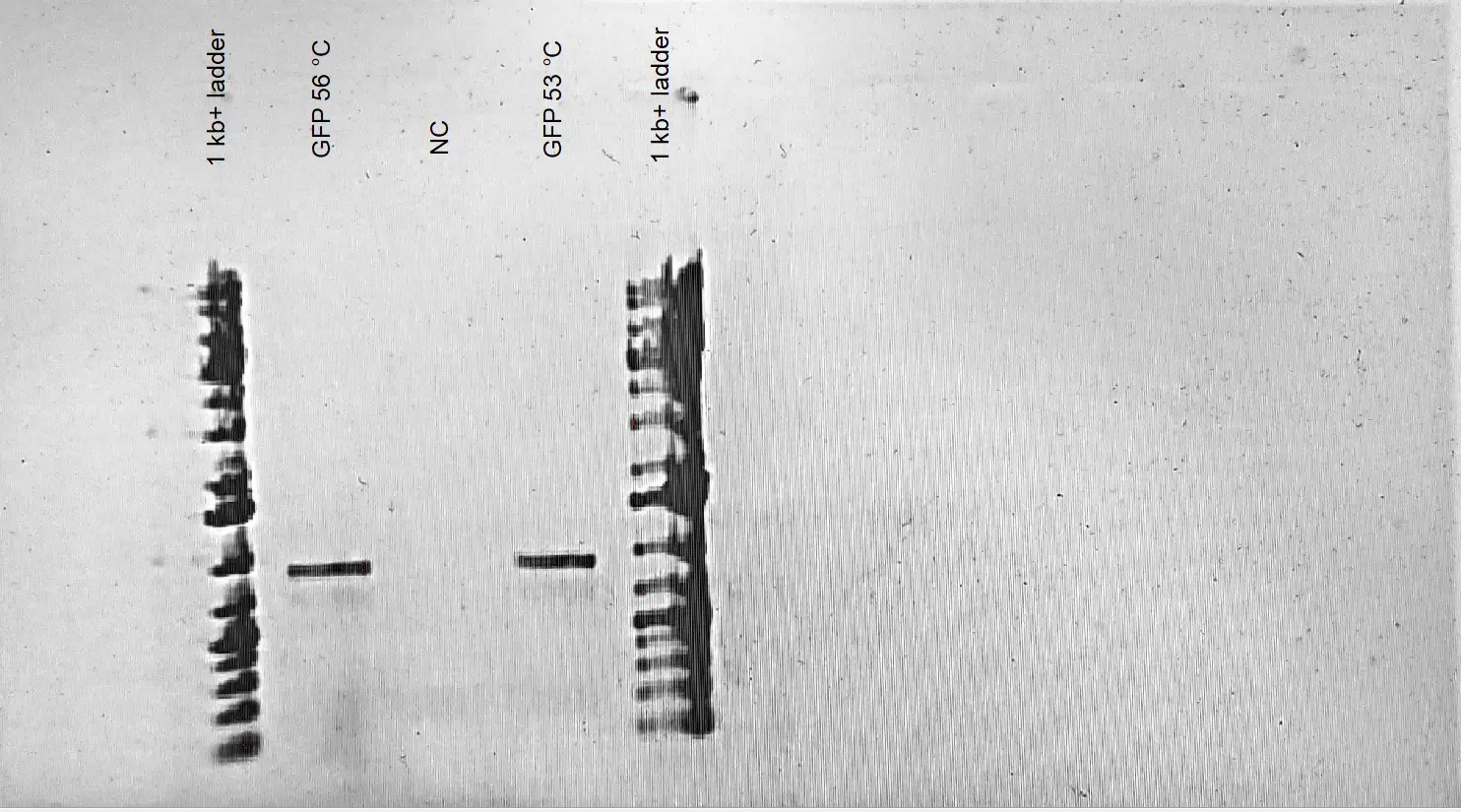
1% Agarose Gel Electrophoresis Results of GFP + B0015 term sequence. Different annealing temperature values are tested in order to determine the annealing temperature specificity of the PCR reaction. The amplification reaction is conducted from the vector BBa_J364001 provided by IGEM Kit Plate #1. A fragment size around ~ 0.9 led to the conclusion that the desired fragment was successfully amplified from the vector. DIfferent annealing temperatures did not lead to any difference in amplification. - We determined that the corresponding GFP + term sequence is successfully amplified from the plasmid BBa_J364001 provided by IGEM.
9
Test transformations of DH5alpha with the Miniprepped IGEM plasmid DNA- Two test transformations were conducted from previously isolated plasmid DNA of the successful transformation reactions in order to confirm that the [DNA] is the main reason for the failed transformation reactions and to test the efficiency of our transformation protocol. 2.5 uL of miniprepped plasmid DNA (BBa_J23100 and BBa_I20270) is added into 50 uL of DH5alpha competent cells. The transformation by chemical competence protocol was exactly followed as described in the experiment protocols document. The cells were pelleted and 100 uL of the recovered cells were plated onto LB+CAM34c plates. The next day, more than 100 colonies were observed in each plate. We concluded that the DNA provided by iGEM might be problematic to conduct successful transformation reactions.
10
PCR Purifications of PBC, HPA fragment #1 and HPA fragment #2- Accidentally, PE Buffer is added into all the tubes containing PCR products. We therefore performed another PCR reaction for PBC and HPA fragment #1. At the end, all PCR products for each different fragment were collected into a microfuge tube and PCR Purification Protocol of Qiagen is followed as described.
-
Final Nanodrop Values:
PBC = ng/μL, HPA #1= 39.9 ng/μL, HPA #2= 37.5 ng/μL
12
Autoclaving More Pipette Tips and Flasks- These were autoclaved in gravity filter cycle #1 (D=30 min, S=15 min)
13
Overlap PCR for HPA fragments #1 and #2-
An alternative assembly method by overlap PCR reactions is done in order to assemble fragments HPA #1 and #2 instead of Gibson. This assembly method relies on 2 PCR reactions : the first one is being done in order to have a fully sized fragment and the second one is being done to specifically amplify this fully sized fragment (https://www.sciencedirect.com/science/article/pii/S2215016119303358)
Rxn #1:
100 ng per 1 kB -> HPA fragment #1 is ~ 2.3 kB -> around 230 ng
-> HPA fragment #2 is ~485 bp -> around 48.5 ng
Nanodrop Values:
HPA fragment #1 = 40 ng/μL
HPA fragment #2 = 37.5 ng/μL
230ng/ 40 ng/μL = 5.75 μL
48.5 ng/37.5 ng/μL = 1.3 μL
Thermocycling conditions for the first reactions are as follows as it is described in the paper: (each amplification step is 135 seconds)Component [Stock] [Final] 1 Reaction Sterile mqWater - - 31.72 μL PCR Buffer 10X 1X 5 μL MgSO4 25 mM 2.5 mM 5 μL dNTPs 25 mM 0.4 mM 0.8 μL Taq Pol 5 U/mL 0.035U/μL 0.35 μL Pfu 2.5 U/mL 0.0032 U/μL 0.08 μL HPA fragment #1 - - 5.75 μL HPA fragment #2 - - 1.3 μL Final volume - - 50.0 μL 
(Hilgarth and Lennigan, 2016) -
Rxn #2:
Thermocycling conditions for the first reactions is altered from the paper’s reaction conditions and are as follows :Component [Stock] [Final] 1 Reaction MM (x2.4) Sterile mqWater - - 33.39 μL 80.15 μL PCR Buffer 10X 1X 5 μL 12 μL MgSO4 25 mM 2.5 mM 5 μL 12 μL dNTPs 25 mM 0.4 mM 0.8 μL 1.92 μL Taq Pol 5 U/mL 0.025 U/μL 0.25 μL 0.6 μL Pfu 2.5 U/mL 0.0027 U/μL 0.056 μL 0.133 μL Fwd primer 10 μM 0.15 μM 0.75 μL 1.8 μL Rev primer 10 μM 0.15 μM 0.75 μL 1.8 μL Mix total - - 46.0 μL 110.4 μL PCR #1 product 4.0 μL - Final volume 50.0 μL
Initial denaturation 30 seconds at 96
Denaturation 15 seconds at 96
Annealing 30 seconds at 58.6
Elongation 135 seconds at 72
Final extension 5 minutes at 72
Hold 10 degrees
Denaturation + Annealing + Elongation steps are repeated for 30 cycles
14
Gel Electrophoresis for Overlapping PCR Reactions + More PCRs + Creating Comp Cells- A single well-isolated colony is inoculated in 5 mL of LB in a test tube. This colony is grown for 7-8 hours with shaking at 37 C.
-
Approximately 125 mL of LB is transferred into a sterile flask and 100 uL of the colony is added into this flask. This flask is placed into an incubator and cells are grown at 20.5 C and at 125 RPM for 15 hours. The next day, OD600 measurements of the cells were done using a spectrophotometer. Cells are grown until OD600 reaches ~ to 0.55. Upon reaching 0.55 absorbance, approximately 30 mL of cells were transferred into 4 different falcon tubes. These are kept in ice for 10 minutes. In the meantime, the centrifuge is pre-chilled to 4 C. Cells are pelleted by centrifuging at 4000 RPM for 10 minutes. Supernatants are poured out into a liquid waste and the cell pellets are resuspended with adding 10 mL of Inoue Buffer into each tube followed by pipetting thoroughly and gentle swirling. Cells are pelleted by centrifuging at 4000 RPM for 10 minutes. Once again, supernatants are removed and cells are resuspended by adding 2.5 mL Inoue Buffer into each tube followed by pipetting thoroughly and gentle swirling. Resulting resuspension is transferred into a 15mL tube and 750 uL of DMSO is added into the solution. The tube is incubated on ice for 10 minutes and the solution is distributed into 750 uL aliquots and these microfuge tubes are kept in -80 freezer
Component [Stock] [Final] 1 Reaction Master Mix (x5) Sterile mqWater - - 16.72 μL 83.6 μL PCR Buffer 10X 1X 2.5 μL 12.5 μL MgSO4 25 mM 2.5 mM 2.5 μL 12.5 μL dNTPs 25 mM 0.4 mM 0.4 μL 2 μL Taq Pol 5 U/mL 0.029 U/μL 0.145 μL 0.725 μL Pfu 2.5 U/mL 0.0032 U/μL 0.032 μL 0.161 μL Fwd Primer 10 μM 0.3 μM 0.75 μL 3.75 μL Rev Primer 10 μM 0.3 μM 0.75 μL 3.75 μL Mix total - - 23.8 μL 119 μL Template (GFP containing plasmid)
- - 1.2 μL - Final volume - - 25.0 μL -
15
GFP PCRs and Test Transformations for the Comp Cells-
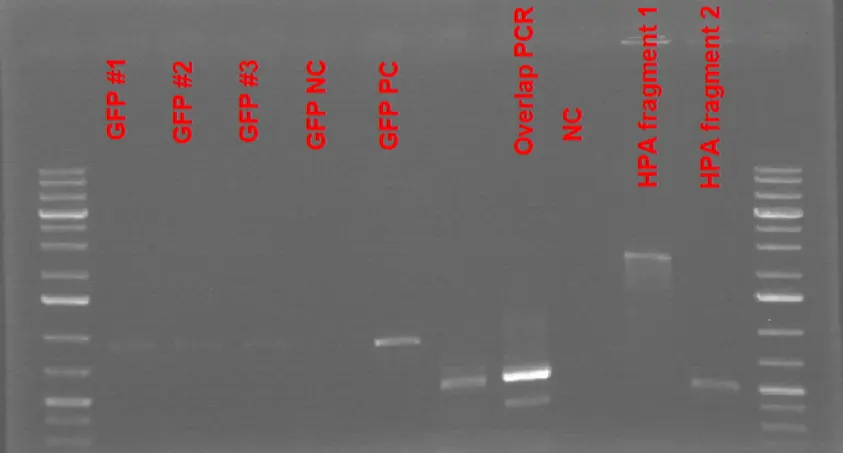
1% Agarose Gel Electrophoresis Results of GFP + B0015 term sequence along with the overlap PCR reaction to assemble 2 HPA fragments. The amplification reaction for GFP is conducted from the vector BBa_J364001 provided by IGEM Kit Plate #1. Very faint bands for GFP amplicons are observed. Overlapping PCR reaction to assemble two HPA fragments did not result in the expected amplification product as the major product seemed to be the same size as the HPA fragment #2.
16
PCR Amplification of GFP-
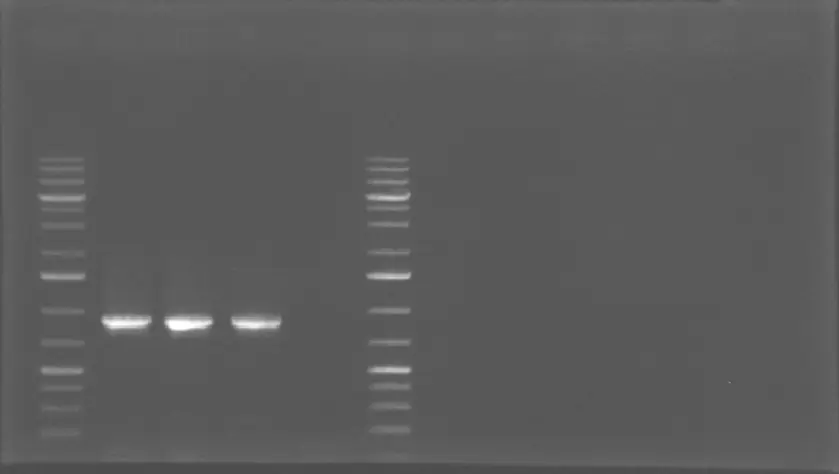
1% Agarose Gel Electrophoresis Results of GFP + B0015 term sequence. The amplification reaction for GFP is conducted from the vector BBa_J364001 provided by IGEM Kit Plate #1. The bands at 900 bp indicate that GFP is successfully amplified with a high [amplicon]
19
PCR Amplification of individual Pbc fragments for the assembly with bam genes + Gibson for individual HPA fragments- PCR reactions were inconclusive due to lack of Taq polymerase stock
20
PCR amplification of the HPA Gibson fragment-
PCR amplification of the full sized HPA fragment from the Gibson reaction is performed. The Gibson reaction is diluted 1:4 with mq water before the PCR
2 ul Gibson product : 8 ul mq waterComponent [Stock] [Final] 1 Reaction Master Mix (x5) Sterile mqWater - - 29.77 μL 148.86 μL PCR Buffer 10X 1X 5 μL 25 μL MgSO4 25 mM 2.5 mM 5 μL 25 μL dNTPs 25 mM 0.4 mM 0.8 μL 4 μL Taq Pol 5 U/mL 0.035 U/μL 0.35 μL 1.75 μL Pfu 2.5 U/mL 0.0039 U/μL 0.078 μL 0.389 μL Fwd Primer 10 μM 0.4 μM 2 μL 10 μL Rev Primer 10 μM 0.4 μM 2 μL 10 μL Mix total - - 45 μL 225 μL Template (Gibson resuspension) - - 5 μL -
Three test tubes for the Gibson PCR :
#1 : 5 uL diluted Gibson product
#2 : 3.5 uL diluted Gibson product + 1.5 uL mqwater
#3 : 1.5 uL diluted Gibson product + 3.5 uL mqwater
#4 : 4 ul overlapping PCR product + 1 ul mqwater
#5 : 5 uL of mqwater is used for the negative control.
Primers : Golden Gate 1 of HPA +Golden Gate 2 of HPA
TMs : 66.7 for fwd , 68.3 for rev -> use 64 C
Initial denat : 2 min at 96
Gel Results :
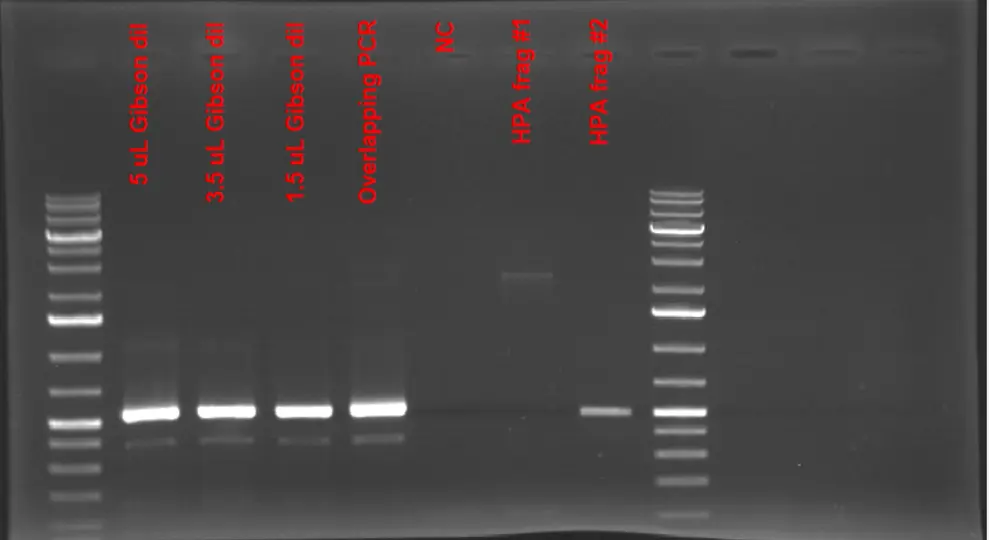
1% Agarose Gel Electrophoresis Results of Gibson Reaction Products used to assemble individual HPA fragments. None of the amplification of the Gibson products did not result in the expected product size of (2.8 kB). HPA fragments # 1 and # are used to confirm that product size of Gibson reaction is different from those fragments
Based on the gel image, we confirmed that the Gibson reaction did not go as planned and no assembly of the two individual fragments to give a full-sized HPA fragment has occurred since we could have had a full-length fragment at the end of the PCR if the Gibson reaction was successful.
Going forward, I decided to amplify the full-length HPA fragment right from the E.coli BL21 genome. This region does contain an internal PaqCI cut site; yet, once it is digested, the ends should only be compatible with one another and not with any other digested fragment. Based on that, our cloning strategy should remain the same although we get the fragment with the internal digestion site.
-
Two reaction tubes + NC tube with the leftoverComponent [Stock] [Final] 1 Reaction Master Mix (x 2.4) Sterile mqWater - - 29.77 μL 71.45 μL PCR Buffer 10X 1X 5 μL 12 μL MgSO4 25 mM 2.5 mM 5 μL 12 μL dNTPs 25 mM 0.4 mM 0.8 μL 1.92 μL Taq Pol 5 U/mL 0.035 U/μL 0.35 μL 0.84 μL Pfu 2.5 U/mL 0.0039 U/μL 0.078 μL 0.187 μL Fwd Primer 10 μM 0.4 μM 2 μL 4.8 μL Rev Primer 10 μM 0.4 μM 2 μL 4.8 μL Mix total - - 45 μL 108 μL Template (cell resuspension) - - 5 μL -
Gel image :
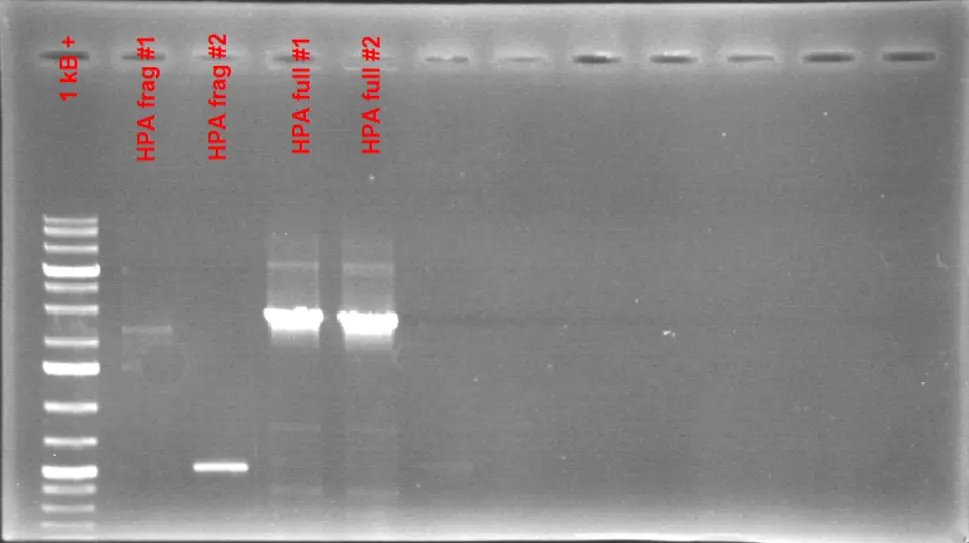
1% Agarose Gel Electrophoresis Results of the full-sized HPA fragment. A colony PCR reaction is conducted to amplify the full-length HPA fragment from E.coli BL21. The resulting product gave a size around 2.8 kB which is the expected length for the full-length HPA fragment.
Based on the gel picture, we can confirm that we got the full-sized HPA fragment. Individual HPA fragments can also be observed on the gel which are in different sizes than the full-length fragment. This fragment can be purified and be used for cloning.
26
PCR amplification of pJUMP27-1A plasmid to introduce BsaI cut sites and PCR Purification of some products-
The low-copy plasmid pJUMP27-1A provided by IGEM is PCR amplified with the tailed-primers containing each BsaI cut sites. The ends of the product should be compatible with the full-sized HPA fragment once digested by BsaI.
One reaction tube + one NCComponent [Stock] [Final] 1 Reaction Master Mix (x 2) Sterile mqWater - - 32.27 μL 65.54 μL PCR Buffer 10X 1X 5 μL 10 μL MgSO4 25 mM 2.5 mM 5 μL 10 μL dNTPs 25 mM 0.4 mM 0.8 μL 1.6 μL Taq Pol 5 U/mL 0.035 U/μL 0.35 μL 0.7 μL Pfu 2.5 U/mL 0.0039 U/μL 0.078 μL 0.156 μL Fwd Primer 10 μM 0.4 μM 2 μL 4 μL Rev Primer 10 μM 0.4 μM 2 μL 4 μL Mix total - - 45 μL 108 μL Template (resuspended plasmid) - - 2.5 μL -
Reaction conditions :
Initial denaturation 120 seconds at 96
Denaturation 15 seconds at 96
Annealing 30 seconds at 52.0
Elongation 170 seconds at 72
Final extension 5 minutes at 72
Hold 10 degrees
Gel Results :

1% Agarose Gel Electrophoresis Results of pJUMP-271A with BsaI ends. pJUMP-271A plasmid provided by IGEM kit Plate #1 is amplified using two primers with BsaI cut sites as tails. The linearized product gave a size around 3 - 3.1 kB which is the expected size of the plasmid
As it can be seen from the gel, the product is around ~ 3- 3.1 kB which is the expected size for the amplicon.
Going forward, the purification of full-sized HPA, GFP and pJUMP27-1A w/ BsaI ends is performed based on the protocol provided by Qiagen PCR Purification Kit.
The products of GFP and pJUMP27-1A are eluted with 40 uL of EB to have more concentrated DNA whereas 50 uL of EB is used to get the HPA fragment.
Final NanoDrop concentrations :
HPA = 100 ng/uL
GFP =
pJUMP27-1A w/ Bsa = 40 ng/uL
28
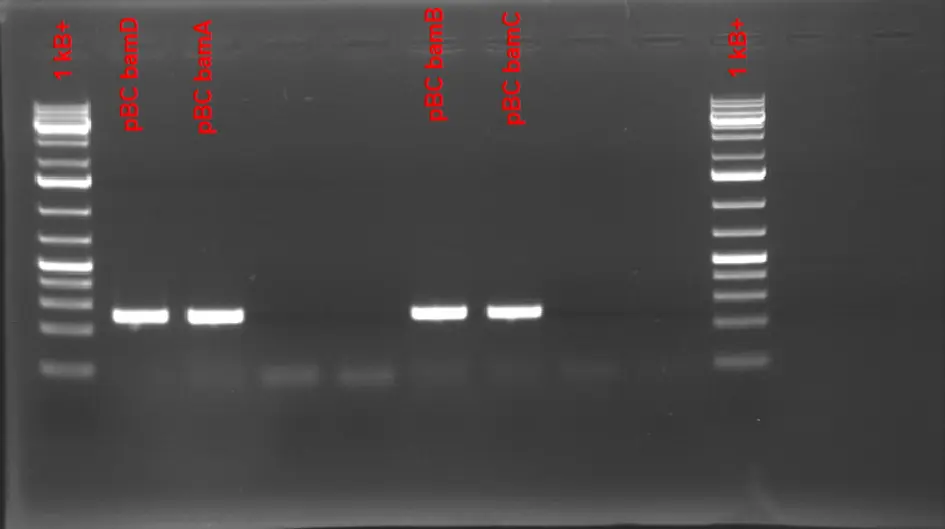
PCR amplification of individual pBC fragments-
E.coli BL21 is PCR amplified with each corresponding primer containing either Golden Gate or Gibson Tails. (Gibson Tails are based on the bam genes)
Reaction conditions:Component [Stock] [Final] 1 Reaction Master Mix (x 2) Sterile mqWater - - 43.16 μL 258.95 μL PCR Buffer 10X 1X 7.5 μL 45 μL MgSO4 25 mM 2.5 mM 7.5 μL 45 μL dNTPs 25 mM 0.4 mM 1.2 μL 7.2 μL Taq Pol 5 U/mL 0.035 U/μL 0.525 μL 3.15 μL Pfu 2.5 U/mL 0.0039 U/μL 0.117 μL 0.7 μL Mix total - - 60 μL 108 μL Fwd Primer 10 μM 0.7 μM 5 μL - Rev Primer 10 μM 0.7 μM 5 μL - Template (resuspended plasmid) - - μL -
Initial denaturation 240 seconds at 96
Denaturation 15 seconds at 96
Annealing 30 seconds at 56.0
Elongation 45 seconds at 72
Final extension 5 minutes at 72
Hold 10 degrees
29
GG Assembly to ligate HPA locus into the target vector pJUMP27-1A with BsaI ends-
For Golden Gate
Full-sized HPA fragment : 2754 bp
Plasmid ~ 3079 bp
80 ng of plasmid -> 40 ng/ul -> 2 ul of plasmid
Molar ratio insert;plasmid -> 80x 2754/3079 = 71.6 ng of insert
2:1 ratio of insert:plasmid -> 71.6 ng x 2 = 143.2 ng
HPA insert -> 100 ng/uL
143.2/100 = 1.43 uL
1 uL GG assembly mix
2 uL T4 Buffer
20 - all = 13.47 uL mq water
3 uL of the reaction product is used for transforming 75 uL of DH5alpha comp cellsComponent [Stock] [Final] Reaction Negative Control Mq Water - - 13.57 uL 15 uL Vector - - 2 uL 2 uL Insert (HPA) - - 1.43 uL - T4 Ligase Buffer 10x 1x 2 uL 2 uL BsaI HF Solution - - 1 uL 1 uL Total - - 20 uL 20 uL - We also transformed 100 uL of Dh5alpha comp cells with IGEM plasmid BBa_J428358
- Transformation for chemical competence procedure is followed
30
GG Assembly to ligate HPA locus into the target vector pJUMP27-1A with BsaI ends- No colonies for the Golden Gate reaction yet the transformation of the IGEM plasmid worked. Either Golden Gate reaction failed or there is something wrong with the kanamycin plates (the stock might be different or kan50 is a lot for a low copy plasmid)
-
Running the components on an agarose gel :
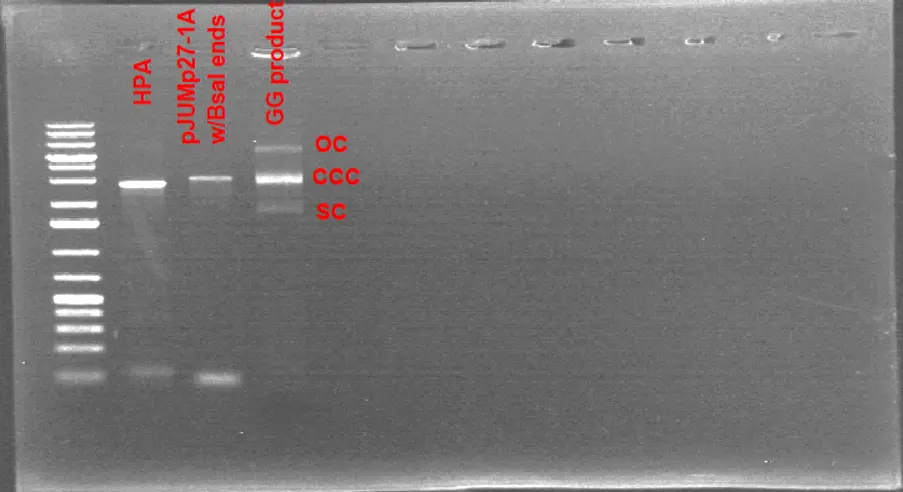
1% Agarose Gel Electrophoresis Results of the Golden Gate Product. As the transformation reactions failed with the corresponding golden gate reaction product, it was run on a gel to compare it with linearized plasmid and HPA fragment. The gel indicates 3 different conformations of a plasmid structure : OC,CCC,SC
It might be possible that the bands corresponding to the Golden Gate Product might be OC, CCC, SC conformations of the plasmid. That would indicate a successful Golden Gate reaction as the linear fragments are circularized
On top agar antibiotic plating is used -> For kan25 (25ug/mL) = Mix 4 uL of Kan 50 mg/mL stock with 196 uL of LB -> Plate 75 uL into each side of the plate both for plating the supernatant and the pellet
31
GG Assembly to ligate HPA locus into the target vector pJUMP27-1A with BsaI ends- Successful colonies observed in both kan25 and kan12.5 plates, the next step is colony PCR to screen the insert
September
5
Colony PCR for screening and patching out the colonies onto new plates-
3 single well isolated colonies from each kan25 and kan12.5 plates were resuspended in 20 uL of mqwater.
2.5 uL of mq water is used for the NCComponent [Stock] [Final] 1 Reaction Master Mix (x 7.2) Sterile mqWater - - 15.40444444 110.912 PCR Buffer 10X 1X 2.5 18 MgSO4 25 mM 2.5 mM 2.5 18 dNTPs 25 mM 0.4 mM 0.4 2.88 Taq Pol 5 U/mL 0.032 U/μL 0.16 1.152 Fwd Primer 10 μM 0.3 μM 0.75 6.48 Rev Primer 10 μM 0.3 μM 0.75 6.48 Mix total - - 22.5 μL 162μL Template (resuspended plasmid) - - 2.5 μL -
Initial denaturation 240 seconds at 96
Denaturation 15 seconds at 96
Annealing 30 seconds at 63.5
Elongation 150 seconds at 72
Final extension 5 minutes at 72On top agar antibiotic plating is used to have the following plates : Kan100 ug/ml, Kan 50ug/ml, Kan 25ug/ml. LB + Kan 50mg/mL combinations were prepared respectively for the corresponding plates : 138 uL LB + 12 uL Kan 50mg/mL stock, 144 uL LB + 6 uL Kan 50mg/mL stock, 147 uL LB + 3 uL Kan 50mg/mL stock
These antibiotic mixtures were then spread onto 3 different LB plates using a glass cell spreader.
Sterile toothpicks were dipped into each resuspension and those were patched out onto each plate
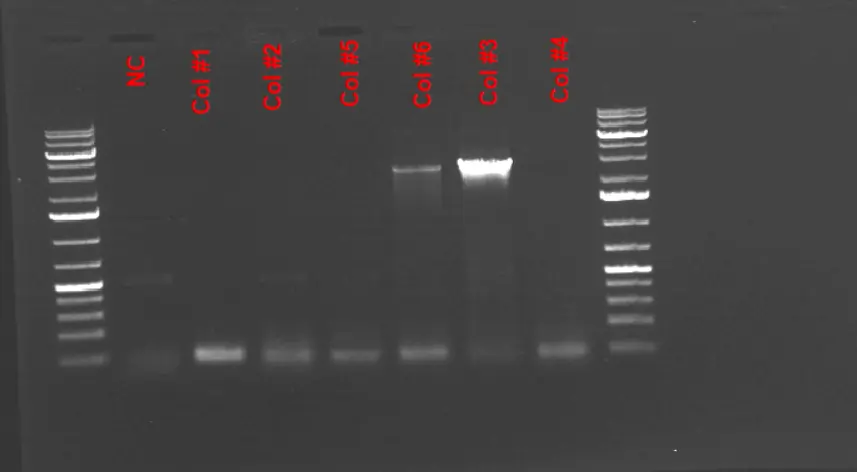
1% Agarose Gel Electrophoresis Results of the Colony PCR to screen for the HPA insert. Successful transformant colonies were diluted in 20 uL of mqwater and a colony PCR is ran to confirm for the presence of HPA insert inside the plasmid.
NOTE : Colonies # 3 and 6 contain the corresponding HPA insert which is around 2.8 kB. We decided to use strain corresponding to colony #3 as it resulted in a thicker band for the insert. -
Component [Stock] [Final] 1 Reaction Master Mix (x 14.4) Sterile mqWater - - 14.90444444 214.624 PCR Buffer 10X 1X 2.5 36 MgSO4 25 mM 2.5 mM 2.5 36 dNTPs 25 mM 0.4 mM 0.4 5.76 Taq Pol 5 U/mL 0.032 U/μL 0.16 2.304 Pfu 2.5 U/mL 0.0036 U/μL 0.03555555556 0.512 Mix total - - 20.5 μL 295.2 μL Fwd Primer 10 μM 0.4 μM 1 2.4 Rev Primer 10 μM 0.4 μM 1 2.4 Mix total - - 22.5 μL - Template (resuspended plasmid) - - 2.5 μL - 
Component [Stock] [Final] 1 Reaction Master Mix (x3) Sterile mqWater - - 17.7 53.1 PCR Buffer 10X 1X 2.5 7.5 MgSO4 50 mM 2.5 mM 1.25 3.75 dNTPs 25 mM 0.4 mM 0.4 1.2 Taq Pol 5 U/mL 0.033 U/μL 0.16 0.5 Fwd Primer 10 μM 0.4 μM 1 3 Rev Primer 10 μM 0.4 μM 1 3 Mix total - - 24 μL 72 Template (resuspended plasmid) - - 1 - 
Component [Stock] [Final] 1 Reaction Master Mix (x 13) Sterile mqWater - - 17.64833333 229.4283333 PCR Buffer 10X 1X 2.5 32.5 MgSO4 25 mM 2.5 mM 1.25 16.25 dNTPs 25 mM 0.4 mM 0.4 5.2 Taq Pol 5 U/mL 0.032 U/μL 0.165 2.145 Pfu 2.5 U/mL 0.0036 U/μL 0.03666666667 0.4766666667 Mix total - - 22 μL 295.2 μL Fwd Primer 10 μM 0.4 μM 1 3.25 Rev Primer 10 μM 0.4 μM 1 3.25 Mix total - - 24 μL - Template (resuspended plasmid) - - 1 μL -