iGEM SORBONNE
NAWI
Improve
Improvement of existing parts
As a part of our partnership with UESTC_BioTech, we decided to help them create a killswitch for their strain of Chlamydomonas. When they evoked their safety concerns for their project, we first thought of our university’s 2020 project, where they had also developed a failsafe for the same reasons. We also thought of making the strain auxotrophic using CRISPR, but after having done some rudimentary research on Sorbonne University (SU) 2020’s project, and not being able to gather much information, we decided the first option would be much more interesting, and thus began our investigative journey.
In exchange, they provided us with a CRISPR system designed to knock out the Psy1 gene in Chlamydomonas reinhardtii, which was shown to decrease chlorophyll production drastically, making colonies completely white [8]. This is of great importance for our project, because chlorophyll is one of the primary sources of unpleasant taste in our cells. Our discussion with local algae-as-food companies revealed that most lab-grown commercial strains lacked chlorophyll for that very reason.
Our killswitch is an improved version of Sorbonne University 2020 iGEM team’s (BBa_K3373014), who were themselves inspired by Munich's 2013 iGEM team. They wanted to address Nuclease A to the plasma membrane by linking it to a transmembranary domain and ER translocation signal peptide. In between the nuclease and transmembranary domain would be a TEV-specific cleavage site. Therefore, under escape conditions, UV-sensitive dimerization could assemble both parts of the TEV protease, liberating the Nuclease A, which would then localize to the nucleus and provoke cell death. In practice, they ran into a few issues that will be discussed later.
Analysis of the parts provided by the 2020 iGEM team was difficult due to dysfunction of the iGEM part registry’s Heroku ApI and the loss of the cambridgeigem.org domain. Therefore, we've manually recreated, reviewed and redesigned both basic and composite parts to fix discrepancies and give complementary informations as to the origin of these parts.
We started by downloading the sequence of their composite part, manually annotating it from the registry’s available info: the annotations coincided with the reported use of the MoClo method. We analyzed these individually to determine the source of each part and created separate parts for each phytobrick, without the restriction and fusion sites, enabling us to assemble the parts by specifying their registry names and scars.
cCA N-peptide secretion signal, taken from carbonic anhydrase 1 of C.reinhardtii, was found in a 2018 article on secretory signal peptides[7]. A new part was created after inferring MoClo sites from the composite part.
Predicted HAP2 transmembranary domain sequence (plus 2 amino acids up and down-stream) was inferred from[2]. It corresponds somewhat to the HAP2 transmembranary domain used in SU 2020's parts.
The 5(4GlySer) linker, inspired by Munich 2013's team, was wrongly annotated. We corrected the annotation, created a basic part for it and deleted the excedentary sequence. The TEV cleavage site was also correct.
The translated sequence of the Nuclease A chain from Staphylococcus aureus's Nuc protein[6] was aligned (using clustal omega) with the translated sequence annotated on both the basic and composite part. It was shown that while both nucleotide sequences differ slightly, translated protein sequences are very similar. We updated both the basic and composite parts with the UniProt reference sequence. The reason for this discrepancy was that 2020’s team deleted a codon in order to remove a BsaI site, we restored it, changing its sequence to the next most-used codon coding the same amino acid.
In both parts, SV40 NLS corresponds to the one published in [3]. But whereas in the basic part there are two repeats of the NLS, in the composite part there is only one. We've adjusted the composite part to reflect the expected result of cloning with the basic part, because tandem NLS have been shown to increase nuclear localisation.
Finally, we verified correct frame positioning between all the parts, and found that there was a one nucleotide frameshift between the TEV cleavage site and NucA. By deleting a nucleotide from the TEV site 3' spacer, we were able to get rid of this frameshift and restore correct translation. Full protein measures 243 amino acids, with a predicted molecular weight of 25536.1 Daltons.
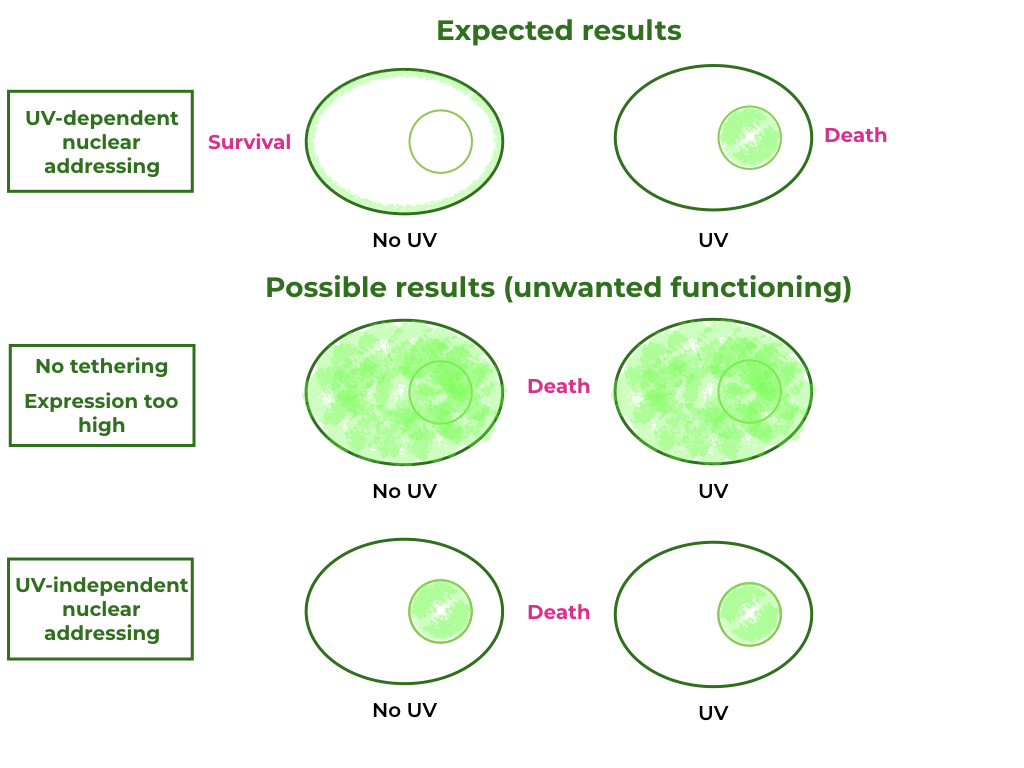
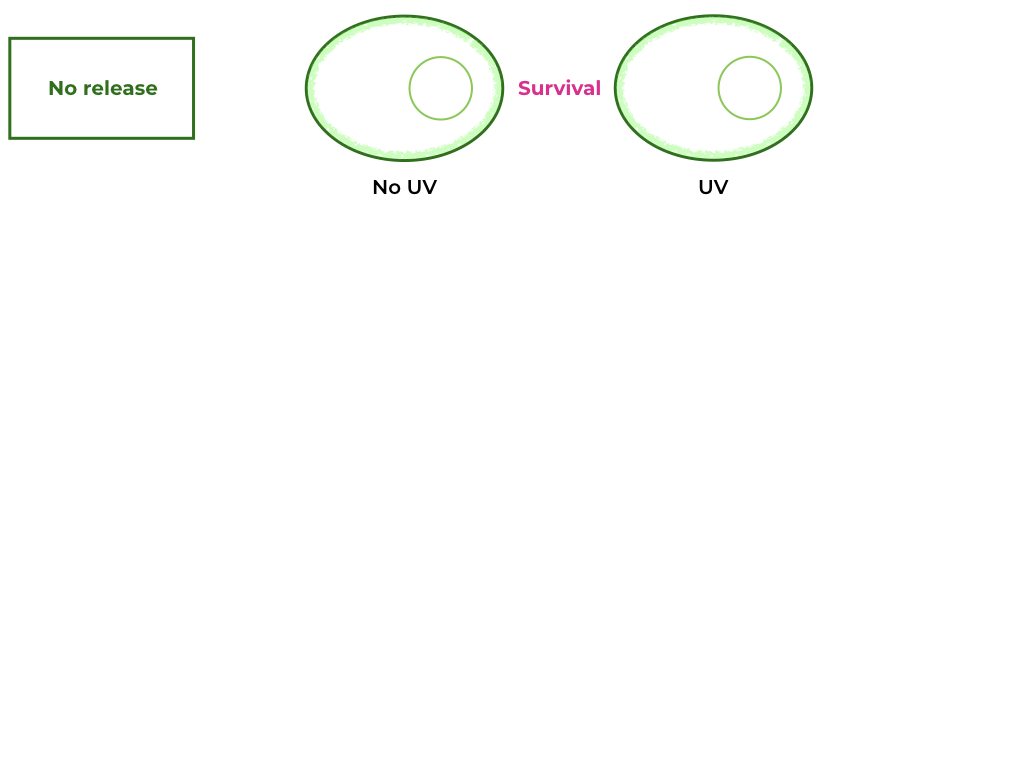
To further our understanding of this killswitch system, additional tests must take place: according to a paper published in 2019 [1], a NLS signal fusioned to a tail anchored membrane protein targeted expression to the inner nuclear envelope in plants. This is a possibility in our construct and therefore would need to be tested. We propose replacing the Nuclease A with a fluorescent protein like GFP, and observing the cells under the fluorescence microscope after induction by UV radiation. Accomplishing this task would allow us to complete Munich 2013’s and Sorbonne 2020’s engineering cycle, by testing and redesigning the circuit accordingly. A more detailed version of this plan can be found at the end of this page.
COP1 and UVR8 in both basic and composite plasmids are identical to the available UniProt refseq for Arabidopsis thaliana (P43254 and Q9FN03, respectively).
Running and xBlast on both C- and N-terminal domains of TEV protease reveals that both have 100% identity with a polyprotein (also named P1 proteinase) from the Tobacco etch virus (UniProtKB P04517).
Proof of concept for split-TEV was demonstrated in a 2006 paper by Wehr et al. (4).Both constructs also have correct reading frames.
Furthermore, Munich 2013’s team’s experimental data showed that their killswitch wasn’t effective, as it reduced viability so much that no cells could be cultivated. Because Nuclease A already has a high activity, we were scared that its overexpression could reduce viability in transformants. By replacing the pSAD terminator with the RbcS-2b terminator, we expect to decrease protein expression 10-fold in Chlamydomonas [5], therefore improving cell viability. Expression could be reduced further by replacing the pSAD promoter with pAR promoter, decreasing expression another 10-fold [5].
This change in expression level would have to be coupled with a UV-sensitivity test, to make sure that our transformants remain sensitive to UV-light-induced cell death. To test the UV-sensitivity without inducing cell death, one could replace the Nuclease A - NLS block with a simple GFP protein, and after UV exposition, it would be possible to differentiate between cells with cytoplasmic GFP and membrane-bound GFP.
As described earlier, one BsaI restriction site found in the Nuclease A CDS, causing us to redesign the CDS in order to remove the restriction site but not alter the amino acid sequence of the produced protein: GAGACC (in frame) becomes GAGACG, as to use the second-most optimal threonine codon (after ACC). Afterwards, no BbsI or BsaI restriction sites were found in either composite or basic parts.
Finally, only one BsaI site was present at the extremities of our composite part; we added a second one (in 5' of our sequence) as to make it resemble the natural result of a level 2 MoClo assembly.
These modifications bring the constructs up to code with the Chlamydomonas reinhardtii MoClo toolkit, meaning that the parts can be used modularly with all available C. reinhardtii MoClo parts.